Lattice Tutorial
The following notebook provides a thorough walkthrough to using the
Lattice class to build up crystal systems.
Lattice Functionality
Variable-dimension crystal structures
Latticesupports the dimensionality ofmBuild, which means that the systems can be in 1D, 2D, or 3D. Replace the necessary vector components with 0 to emulate the dimensionality of interest.
Multicomponent crystals
Latticecan support an indefinite amount of lattice points in its data structure.The repeat cell can be as large as the user defines useful.
The components that occupy the lattice points are
mbuild.Compoundobjects.This allows the user to build any system since compounds are only a representation of matter in this design.
Molecular crystals, coarse grained, atomic, even alloy crystal structures are all supported
Triclinic Lattices
With full support for triclinic lattices, any crystal structure can technically be developed.
Either the user can provide the lattice parameters, or if they know the vectors that span the unit cell, that can also be provided.
Generation of lattice structure from crystallographic index file (CIF) formats
Please also see the Load files section for other ways to load files.
IN PROGRESS Template recipes to generate common crystal structures (FCC, BCC, HEX, etc)
This is currently being developed and will be released relatively shortly
To generate these structures currently, the user needs to know the lattice parameters or lattice vectors that define these units.
Lattice Data Structure Introduction
Below we will explore the relevant data structures that are attributes
of the Lattice class. This information will be essential to build
desired crystal structures.
To begin, we will call the python help() method to observe the
parameters and attributes of the Lattice class.
import mbuild
help(mbuild.Lattice)
As we can see, there are quite a few attributes and parameters that make
up this class. There are also a lot of inline examples as well. If you
ever get stuck, remember to use the python built-in help() method!
Lattice.lattice_spacing
This data structure is a (3,) array that details the lengths of the repeat cell for the crystal. You can either use a
numpyarray object, or simply pass in a list andLatticewill handle the rest. Remember thatmBuild’s units of length are in nanometers [nm]. You must pass in all three lengths, even if they are all equivalent. These are the lattice parameters \(a, b, c\) when viewing crystallographic information.For Example:
lattice_spacing = [0.5, 0.5, 0.5]
Lattice.lattice_vectors
lattice_vectorsis a 3x3 array that defines the vectors that encapsulate the repeat cell. This is an optional value that the user can pass in to define the cell. Either this must be passed in, or the 3 Bravais angles of the cell from the lattice parameters must be provided. If neither is passed in, the default value are the vectors that encase a cubic lattice.Note
Most users will not have to use these to build their lattice structure of interest. It will usually be easier for the users to provide the 3 Bravais angles instead. If the user then wants the vectors, the
Latticeobject will calculate them for the user.For example: Cubic Cell
lattice_vectors = [[1, 0, 0], [0, 1, 0], [0, 0, 1]]
Lattice.angles
anglesis a (3,) array that defines the three Bravais angles of the lattice. Commonly referred to as \(\alpha, \beta, \gamma\) in the definition of the lattice parameters.For example: Cubic Cell
angles = [90, 90, 90]
Lattice.lattice_points
lattice_pointscan be the most common source of confusion when creating a crystal structure. In crystallographic terms, this is the minimum basis set of points in space that define where the points in the lattice exist. This requires that the user does not over define the system.Note
MIT’s OpenCourseWare has an excellent PDF for more information here
The other tricky issue that can come up is the data structure itself.
lattice_pointsis a dictionary where thedict.keyitems are thestringid’s for each basis point. Thedict.valuesitems are a nested list of fractional coordinates of the unique lattice points in the cell. If you have the sameCompoundat multiple lattice_points, it is easier to put all those coordinates in a nested list under the samekeyvalue. Two examples will be given below, both FCC unit cells, one with all the same id, and one with unique ids for each lattice_point.For Example: FCC All Unique
lattice_points = {'A' : [[0, 0, 0]], 'B' : [[0.5, 0.5, 0]], 'C' : [[0.5, 0, 0.5]], 'D' : [[0, 0.5, 0.5]] }
For Example: FCC All Same
lattice_points = {'A' : [[0, 0, 0], [0.5, 0.5, 0], [0.5, 0, 0.5], [0, 0.5, 0.5]] }
Lattice Public Methods
The Lattice class also contains methods that are responsible for
applying Compounds to the lattice points, with user defined cell
replications in the x, y, and z directions.
Lattice.populate(compound_dict=None, x=1, y=1, z=1)
This method uses the
Latticeobject to placeCompoundsat the specifiedlattice_points. There are 4 optional inputs for this class.compound_dictinputs another dictionary that defines a relationship between thelattice_pointsand theCompoundsthat the user wants to populate the lattice with. Thedict.keysof this dictionary must be the same as thekeysin thelattice_pointsdictionary. However, for thedict.itemsin this case, theCompoundthat the user wants to place at that lattice point(s) will be used. An example will use the FCC examples from above. They have been copied below:For Example: FCC All Unique
lattice_points = {'A' : [[0, 0, 0]], 'B' : [[0.5, 0.5, 0]], 'C' : [[0.5, 0, 0.5]], 'D' : [[0, 0.5, 0.5]] } # compound dictionary a = mbuild.Compound(name='A') b = mbuild.Compound(name='B') c = mbuild.Compound(name='C') d = mbuild.Compound(name='D') compound_dict = {'A' : a, 'B' : b, 'C' : c, 'D' : d}
For Example: FCC All Same
lattice_points = {'A' : [[0, 0, 0], [0.5, 0.5, 0], [0.5, 0, 0.5], [0, 0.5, 0.5]] } # compound dictionary a = mbuild.Compound(name='A') compound_dict = {'A' : a}
Example Lattice Systems
Below contains some examples of homogeneous and heterogeneous 2D and 3D
lattice structures using the Lattice class.
Simple Cubic (SC)
Polonium
import mbuild as mb
import numpy as np
# define all necessary lattice parameters
spacings = [0.3359, 0.3359, 0.3359]
angles = [90, 90, 90]
points = [[0, 0, 0]]
# define lattice object
sc_lattice = mb.Lattice(lattice_spacing=spacings, angles=angles, lattice_points={'Po' : points})
# define Polonium Compound
po = mb.Compound(name='Po')
# populate lattice with compounds
po_lattice = sc_lattice.populate(compound_dict={'Po' : po}, x=2, y=2, z=2)
# visualize
po_lattice.visualize()
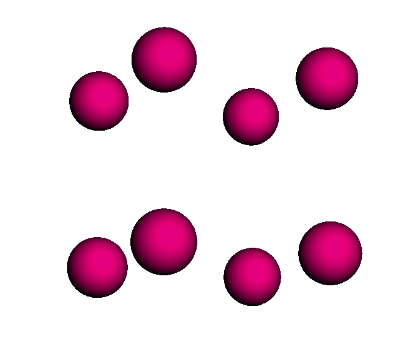
Polonium simple cubic (SC) structure
Body-centered Cubic (BCC)
CsCl
import mbuild as mb
import numpy as np
# define all necessary lattice parameters
spacings = [0.4123, 0.4123, 0.4123]
angles = [90, 90, 90]
point1 = [[0, 0, 0]]
point2 = [[0.5, 0.5, 0.5]]
# define lattice object
bcc_lattice = mb.Lattice(lattice_spacing=spacings, angles=angles, lattice_points={'A' : point1, 'B' : point2})
# define Compounds
cl = mb.Compound(name='Cl')
cs = mb.Compound(name='Cs')
# populate lattice with compounds
cscl_lattice = bcc_lattice.populate(compound_dict={'A' : cl, 'B' : cs}, x=2, y=2, z=2)
# visualize
cscl_lattice.visualize()
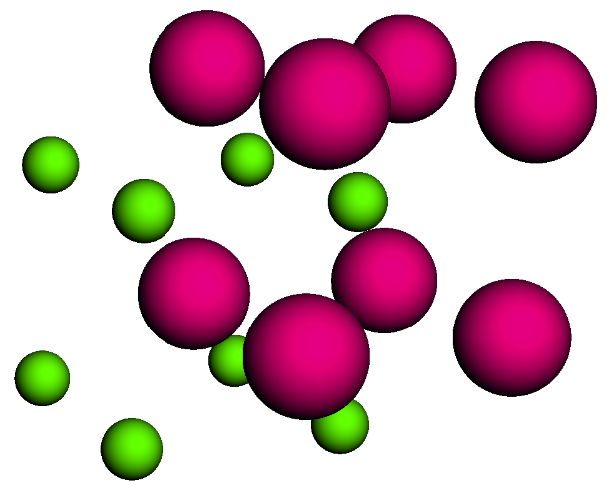
CsCl body-centered cubic (BCC) structure
Face-centered Cubic (FCC)
Cu
import mbuild as mb
import numpy as np
# define all necessary lattice parameters
spacings = [0.36149, 0.36149, 0.36149]
angles = [90, 90, 90]
points = [[0, 0, 0], [0.5, 0.5, 0], [0.5, 0, 0.5], [0, 0.5, 0.5]]
# define lattice object
fcc_lattice = mb.Lattice(lattice_spacing=spacings, angles=angles, lattice_points={'A' : points})
# define Compound
cu = mb.Compound(name='Cu')
# populate lattice with compounds
cu_lattice = fcc_lattice.populate(compound_dict={'A' : cu}, x=2, y=2, z=2)
# visualize
cu_lattice.visualize()

Cu face-centered cubic (FCC) structure
Diamond (Cubic)
Si
import mbuild as mb
import numpy as np
# define all necessary lattice parameters
spacings = [0.54309, 0.54309, 0.54309]
angles = [90, 90, 90]
points = [[0, 0, 0], [0.5, 0.5, 0], [0.5, 0, 0.5], [0, 0.5, 0.5],
[0.25, 0.25, 0.75], [0.25, 0.75, 0.25], [0.75, 0.25, 0.25], [0.75, 0.75, 0.75]]
# define lattice object
diamond_lattice = mb.Lattice(lattice_spacing=spacings, angles=angles, lattice_points={'A' : points})
# define Compound
si = mb.Compound(name='Si')
# populate lattice with compounds
si_lattice = diamond_lattice.populate(compound_dict={'A' : si}, x=2, y=2, z=2)
# visualize
si_lattice.visualize()
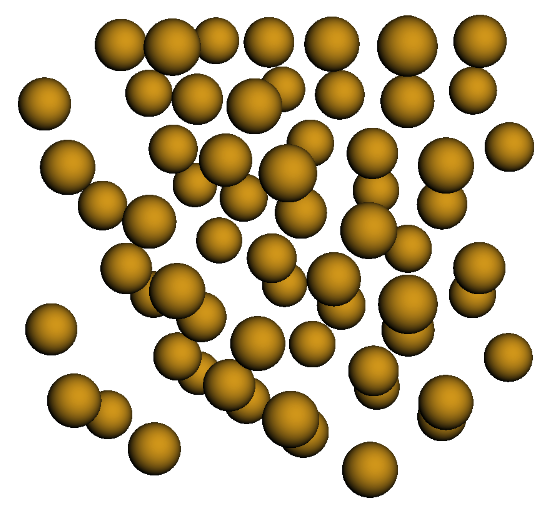
Si diamond (Cubic) structure
Graphene (2D)
C
import mbuild as mb
import numpy as np
# define all necessary lattice parameters
spacings = [0.246, 0.246, 0.335]
angles = [90, 90, 120]
points = [[0, 0, 0], [1/3, 2/3, 0]]
# define lattice object
graphene_lattice = mb.Lattice(lattice_spacing=spacings, angles=angles, lattice_points={'A' : points})
# define Compound
c = mb.Compound(name='C')
# populate lattice with compounds
graphene = graphene_lattice.populate(compound_dict={'A' : c}, x=5, y=5, z=1)
# visualize
graphene.visualize()
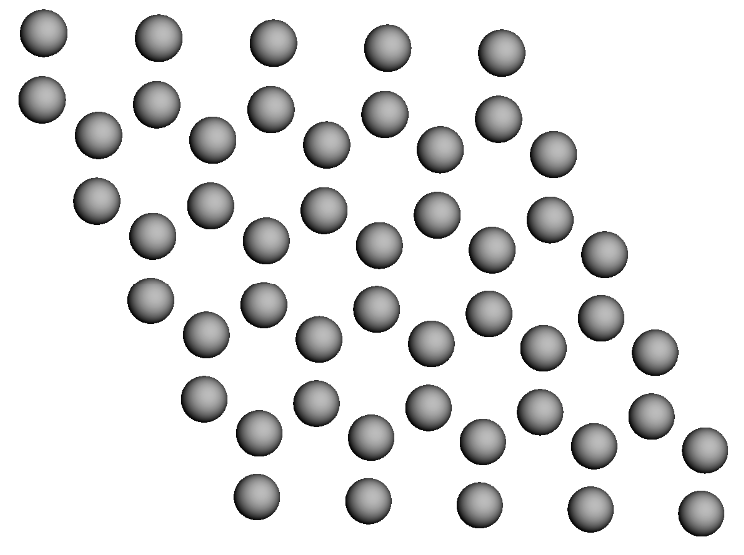
Graphene (2D) structure