Building a Simple Alkane
The purpose of this tutorial is to demonstrate the construction of an alkane polymer and provide familiarity with many of the underlying functions in mBuild. Note that a robust polymer construction recipe already exists in mBuild, which will also be demonstrated at the end of the tutorial.
Setting up the monomer
The first step is to construct the basic repeat unit for the alkane, i.e., a \(CH_2\) group, similar to the construction of the \(CH_3\) monomer in the prior methane tutorial. Rather than importing the coordinates from a pdb file, as in the previous example, we will instead explicitly define them in the class. Recall that distance units are nm in mBuild.
import mbuild as mb
class CH2(mb.Compound):
def __init__(self):
super(CH2, self).__init__()
# Add carbon
self.add(mb.Particle(name='C', pos=[0,0,0]), label='C[$]')
# Add hydrogens
self.add(mb.Particle(name='H', pos=[-0.109, 0, 0.0]), label='HC[$]')
self.add(mb.Particle(name='H', pos=[0.109, 0, 0.0]), label='HC[$]')
# Add bonds between the atoms
self.add_bond((self['C'][0], self['HC'][0]))
self.add_bond((self['C'][0], self['HC'][1]))
# Add ports anchored to the carbon
self.add(mb.Port(anchor=self[0]), label='up')
self.add(mb.Port(anchor=self[0]), label='down')
# Move the ports approximately half a C-C bond length away from the carbon
self['up'].translate([0, -0.154/2, 0])
self['down'].translate([0, 0.154/2, 0])
monomer = CH2()
monomer.visualize(show_ports=True)
This configuration of the monomer is not a particularly realistic conformation. One could use this monomer to construct a polymer and then apply an energy minimization scheme, or, as we will demonstrate here, we can use mBuild’s rotation commands to provide a more realistic starting point.
Below, we use the same basic script, but now apply a rotation to the hydrogen atoms. Since the hydrogens start 180° apart and we know they should be ~109.5° apart, each should be rotated half of the difference closer to each other around the y-axis. Note that the rotation angle is given in radians. Similarly, the ports should be rotated around the x-axis by the same amount so that atoms can be added in a realistic orientation.
import numpy as np
import mbuild as mb
class CH2(mb.Compound):
def __init__(self):
super(CH2, self).__init__()
# Add carbon
self.add(mb.Particle(name='C', pos=[0,0,0]), label='C[$]')
# Add hydrogens
self.add(mb.Particle(name='H', pos=[-0.109, 0, 0.0]), label='HC[$]')
self.add(mb.Particle(name='H', pos=[0.109, 0, 0.0]), label='HC[$]')
# Rotate the hydrogens
theta = 0.5 * (180 - 109.5) * np.pi / 180
#mb.rotate(self['HC'][0], theta, around=[0, 1, 0])
#mb.rotate(self['HC'][1], -theta, around=[0, 1, 0])
self['HC'][0].rotate( theta, around=[0, 1, 0])
self['HC'][1].rotate(-theta, around=[0, 1, 0])
# Add bonds between the atoms
self.add_bond((self['C'][0], self['HC'][0]))
self.add_bond((self['C'][0], self['HC'][1]))
# Add the ports and appropriately rotate them
self.add(mb.Port(anchor=self[0]), label='up')
self['up'].translate([0, -0.154/2, 0])
self['up'].rotate(theta, around=[1, 0, 0])
self.add(mb.Port(anchor=self[0]), label='down')
self['down'].translate([0, 0.154/2, 0])
self['down'].rotate(-theta, around=[1, 0, 0])
monomer = CH2()
monomer.visualize(show_ports=True)
Defining the polymerization class
With a basic monomer construct, we can now construct a polymer by
connecting the ports together. Here, we first instantiate one instance
of the CH2 class as 1ast_monomer, then use the clone function to
make a copy. The force_overlap() function is used to connect the
'up' port from current_monomer to the 'down' port of
last_mononer.
class AlkanePolymer(mb.Compound):
def __init__(self):
super(AlkanePolymer, self).__init__()
last_monomer = CH2()
self.add(last_monomer)
for i in range(3):
current_monomer = CH2()
mb.force_overlap(move_this=current_monomer,
from_positions=current_monomer['up'],
to_positions=last_monomer['down'])
self.add(current_monomer)
last_monomer = current_monomer
polymer = AlkanePolymer()
polymer.visualize(show_ports=True)
Visualization of this structure demonstrates a problem; the polymer curls up on itself. This is a result of the fact that ports not only define the location in space, but also an orientation. This can be trivially fixed, by rotating the down port 180° around the y-axis.
We can also add a variable chain_length both to the for loop and
init that will allow the length of the polymer to be adjusted when
the class is instantiated.
import numpy as np
import mbuild as mb
class CH2(mb.Compound):
def __init__(self):
super(CH2, self).__init__()
# Add carbons and hydrogens
self.add(mb.Particle(name='C', pos=[0,0,0]), label='C[$]')
self.add(mb.Particle(name='H', pos=[-0.109, 0, 0.0]), label='HC[$]')
self.add(mb.Particle(name='H', pos=[0.109, 0, 0.0]), label='HC[$]')
# rotate hydrogens
theta = 0.5 * (180 - 109.5) * np.pi / 180
self['HC'][0].rotate(theta, around=[0, 1, 0])
self['HC'][1].rotate(-theta, around=[0, 1, 0])
# Add bonds between the atoms
self.add_bond((self['C'][0], self['HC'][0]))
self.add_bond((self['C'][0], self['HC'][1]))
# Add ports
self.add(mb.Port(anchor=self[0]), label='up')
self['up'].translate([0, -0.154/2, 0])
self['up'].rotate(theta, around=[1, 0, 0])
self.add(mb.Port(anchor=self[0]), label='down')
self['down'].translate([0, 0.154/2, 0])
self['down'].rotate(np.pi, [0, 1, 0])
self['down'].rotate(-theta, around=[1, 0, 0])
class AlkanePolymer(mb.Compound):
def __init__(self, chain_length=1):
super(AlkanePolymer, self).__init__()
last_monomer = CH2()
self.add(last_monomer)
for i in range (chain_length-1):
current_monomer = CH2()
mb.force_overlap(move_this=current_monomer,
from_positions=current_monomer['up'],
to_positions=last_monomer['down'])
self.add(current_monomer)
last_monomer=current_monomer
polymer = AlkanePolymer(chain_length=10)
polymer.visualize(show_ports=True)
Using mBuild’s Polymer Class
mBuild provides a prebuilt class to perform this basic
functionality. Since it is designed to be more general, it takes as an
argument not just the replicates (n), sequence (‘A’ for a single monomer or ‘AB’ for two different monomers).
Then, it binds them together by removing atom/bead via specifying its index number (indices).
A graphical description of the polymer builder creating ports, then bonding them together is provided below.
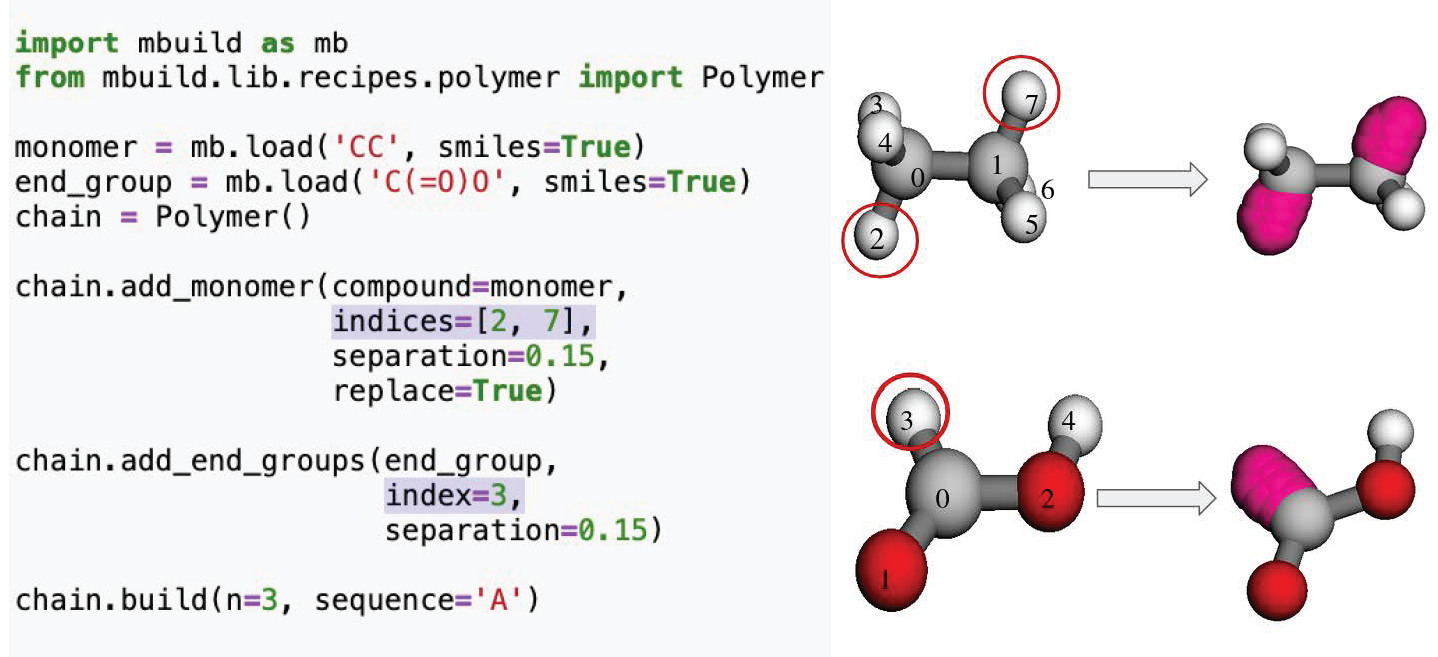
Polymer builder class example. This shows how to define the atoms, which are replaced with ports. The ports are then bonded together between the monomers. Additionally, these ports can be utilized for adding different end groups moieties to the polymer.
Note
The port locations may be critical to ensure the molecule is not overlapping when it is built.
Building a Simple Hexane
A simple hexane molecule is built using mBuild’s packaged polymer builder.
This is done by loading a methane molecule via a SMILES string.
The indices are explicitly selected, so the molecule builds out in the proper directions and does not overlap.
import mbuild as mb
from mbuild.lib.recipes.polymer import Polymer
comp = mb.load('C', smiles=True) # mBuild compound of the monomer unit
chain = Polymer()
chain.add_monomer(compound=comp,
indices=[1, -2],
separation=.15,
replace=True)
chain.build(n=6, sequence='A')
Using Multiple Monomers and Capping the Ends of a Polymer
This example uses methyl ether and methane monomers to build a polymer, capping it with fluorinated and alcohol end groups.
The monomers are combined together in the ‘AB’ sequence two times (n=2), which means the polymer will contain 2 of each monomer (ABAB).
The end groups are added via the add_end_groups attribute, specifying the atom to use (index), the distance of the bond (separation),
the location of each end group (label), and if the tail end group is duplicated to the head of the polymer (duplicate).
The indices are explicitly selected, so the molecule builds out in the proper directions and does not overlap.
from mbuild.lib.recipes.polymer import Polymer
import mbuild as mb
comp_1 = mb.load('C', smiles=True)
comp_2 = mb.load('COC', smiles=True)
chain = Polymer()
chain.add_monomer(compound=comp_1,
indices=[1, -1],
separation=.15,
replace=True)
chain.add_monomer(compound=comp_2,
indices=[3, -1],
separation=.15,
replace=True)
chain.add_end_groups(mb.load('O',smiles=True), # Capping off this polymer with an Alcohol
index=1,
separation=0.15, label="head", duplicate=False)
chain.add_end_groups(mb.load('F',smiles=True), # Capping off this polymer with a Fluorine
index=1,
separation=0.18, label="tail", duplicate=False)
chain.build(n=2, sequence='AB')
chain.visualize(show_ports=True)
Building a System of Alkanes
A system of alkanes can be constructed by simply cloning the polymer
constructed above and translating and/or rotating the alkanes in space.
mBuild provides many routines that can be used to create different
patterns, to which the polymers can be shifted.
comp = mb.load('C', smiles=True) # mBuild compound of the monomer unit
polymer = Polymer()
polymer.add_monomer(compound=comp,
indices=[1, -2],
separation=.15,
replace=True)
polymer.build(n=10, sequence='A')
# the pattern we generate puts points in the xy-plane, so we'll rotate the polymer
# so that it is oriented normal to the xy-plane
polymer.rotate(np.pi/2, [1, 0, 0])
# define a compound to hold all the polymers
system = mb.Compound()
# create a pattern of points to fill a disk
# patterns are generated between 0 and 1,
# and thus need to be scaled to provide appropriate spacing
pattern_disk = mb.DiskPattern(50)
pattern_disk.scale(5)
# now clone the polymer and move it to the points in the pattern
for pos in pattern_disk:
current_polymer = mb.clone(polymer)
current_polymer.translate(pos)
system.add(current_polymer)
system.visualize()
Other patterns can be used, e.g., the Grid3DPattern. We can also use
the rotation commands to randomize the orientation.
import random
comp = mb.load('C', smiles=True)
polymer = Polymer()
polymer.add_monomer(compound=comp,
indices=[1, -2],
separation=.15,
replace=True)
polymer.build(n=10, sequence='A')
system = mb.Compound()
polymer.rotate(np.pi/2, [1, 0, 0])
pattern_disk = mb.Grid3DPattern(5, 5, 5)
pattern_disk.scale(8.0)
for pos in pattern_disk:
current_polymer = mb.clone(polymer)
for around in [(1, 0, 0), (0, 1, 0), (0, 0, 1)]: # rotate around x, y, and z
current_polymer.rotate(random.uniform(0, np.pi), around)
current_polymer.translate(pos)
system.add(current_polymer)
system.visualize()
mBuild also provides an interface to PACKMOL, allowing the
creation of a randomized configuration.
comp = mb.load('C', smiles=True) # mBuild compound of the monomer unit
polymer = Polymer()
polymer.add_monomer(compound=comp,
indices=[1, -2],
separation=.15,
replace=True)
polymer.build(n=5, sequence='A')
system = mb.fill_box(polymer, n_compounds=100, overlap=1.5, box=[10,10,10])
system.visualize()
Variations
Rather than a linear chain, the Polymer class we wrote can be easily
changed such that small perturbations are given to each port. To avoid
accumulation of deviations from the equilibrium angle, we will clone an
unperturbed monomer each time (i.e., monomer_proto) before applying
a random variation.
We also define a variable delta, which will control the maximum
amount of perturbation. Note that large values of delta may result
in the chain overlapping itself, as mBuild does not currently
include routines to exclude such overlaps.
import mbuild as mb
import random
class AlkanePolymer(mb.Compound):
def __init__(self, chain_length=1, delta=0):
super(AlkanePolymer, self).__init__()
monomer_proto = CH2()
last_monomer = CH2()
last_monomer['down'].rotate(random.uniform(-delta,delta), [1, 0, 0])
last_monomer['down'].rotate(random.uniform(-delta,delta), [0, 1, 0])
self.add(last_monomer)
for i in range(chain_length-1):
current_monomer = mb.clone(monomer_proto)
current_monomer['down'].rotate(random.uniform(-delta,delta), [1, 0, 0])
current_monomer['down'].rotate(random.uniform(-delta,delta), [0, 1, 0])
mb.force_overlap(move_this=current_monomer,
from_positions=current_monomer['up'],
to_positions=last_monomer['down'])
self.add(current_monomer)
last_monomer=current_monomer
polymer = AlkanePolymer(chain_length = 200, delta=0.4)
polymer.visualize()